
什么是雷特综合征《下篇》
2017年12月06日 上海丫丫爸基因的突变
有了上面关于基因表达知识基础,现在理解基因突变对最终表现的影响就比较简单了。我们继续看MECP2基因,考虑不同的突变情况。
假设基因的碱基排列是ATGAAGCGC……,现在基因发生了突变,那么可能有下面几种情况:
1.某个碱基突变了,但是不影响氨基酸。例如第9个碱基从C突变成G,那么所在的密码子就从CGC变成了CGG。尽管碱基发生了变化,但是不管CGC和CGG,最终都会被翻译成精氨酸。因此这种突变是不影响蛋白质表达的,称为同义突变或者沉默突变(Silence Mutation),沉默突变是不会影响最终表型的。
2.某个碱基突变了,导致原先编码的氨基酸变成了另外一种氨基酸。例如第7个碱基从C突变成G,那么所在的密码子就从CGC变成为GGC,会导致应翻译的精氨酸变成了甘氨酸,这种突变称为错义突变(Missense Mutation)。
3.某个碱基突变了,导致原先编码的氨基酸变成了结束符号(Stop)。例如第4个碱基从A突变成T,那么所在的密码子就从AAG变称为TAG,会导致应翻译的赖氨酸无法翻译,并且翻译由此结束,后继的密码子将不再被翻译,这种突变称为无义突变(Nonsense Mutation)。
4.某个位置插入了一个多余的碱基,或者一个碱基丢失,导致DNA序列整体偏移,例如第4个碱基A丢失,那么所在的密码子就从AAG变成AGC,并且后面的翻译都会出现问题,这种突变称为移码突变(Frameshift Mutation)。
5.其它复杂情况,例如大片段的碱基丢失或碱基插入,碱基重复,多个碱基突变,等等。
基因型和表型的关系
有了上面对MECP2基因各个功能区的分析,以及对于基因表达的介绍,那么到这里大家很自然的会想:不同的基因突变是否会影响最终的表现?也就是基因型和表型之间是否会有比较确定的对应关系?
更明确的:
1.是否错义突变会“好于”无义突变?
2.是否突变位置靠后的无义突变“好于”突变位置靠前的无义突变?
3.是否在不重要的地方的错义突变“好于”在重要地方(e.g. MBD/TRD)的错义突变?
4.是否氨基酸特性差不多的错义突变会“好于”氨基酸特性相差很多的错义突变?
等等。
答案是,似乎是,但不肯定。在Percy的2017年欧盟雷特大会上引用的文献也提到:“R133C,R294X,R306C,以及3’truncations相对于R106W,R168X,R255X,R270X,splice site和del/ins这些来说,表现要更好(轻)一些”[10]。
这里先稍微解释一下这些突变的具体意思,详细的解读方法会在后面讲解。比如说,R133C的意思就是,在对应MeCP2蛋白质的第133个氨基酸的地方,因为基因突变导致R(Arg,精氨酸)突变成为 C(Cys,半胱氨酸),这是一个错义突变。而R168X的意思是,在第168个氨基酸的地方,R(精氨酸)突变成为终止码Stop,这是一个无义突变。del/ins的意思是大片段的基因更改,从而产生很多不可预知的结果,通常也会提前终止。而3’truncations,splice site这些后面再细说。
因此,R133C这个突变,因为是错义突变,且突变是从亲水的碱性氨基酸Arg变成亲水的中性氨基酸Cyc,差别不是特别大,所以表现会好一些。R294X和R306C突变的位置在TRD区域(207~310)中很靠后了,所以表现也会好一些。而R106W,虽然是错义突变,但是突变是从亲水的碱性氨基酸Arg变成疏水的氨基酸Trp,差别大一些,所以表现会相对差一些。R168X、R255X和R270X都是无义突变,且都发生在MBD或TRD靠前一些的地方,蛋白质编码提前终止,因此表现也会差一些。
此外,有文献[11]表明,有2例语言保留型雷特综合征患者的突变发生在TRD区域之后。这里可以理解为,虽然没有能生成正确的MeCP2蛋白质,但是生成的蛋白质包含有完整的MBD和TRD两个区域,因此表现会相对较好一些(语言保留型)。
但是,需要说明的是,这种相对“好一些”或“差一些”的表现和基因并没有统计学意义,一个原因是样本量太少,另一个原因是还需要考虑X染色体失活(XCI)这个因素。
X染色体失活和表型的关系
回到前面回顾的生物学知识,人类一共有23对染色体,其中有一对性染色体,女性为XX,男性为XY。尽管叫“性”染色体,但是X和Y却大不相同。Y染色体确实可以称为“性”染色体,因为其只包含和男性相关表达的少量基因,而X染色体却不是,它含的基因要多很多,而且有很多对人的生长发育很重要的基因。
根据基因表达的理论,在人体内DNA转录到mRNA再翻译成蛋白质……这样的操作是一直进行的。因此,女性会随机“沉默”一条X染色体,这样才能保证只有1条X染色体上的基因得到表达——否则的话就会有双倍的基因表达,而通常过度的基因表达对人体也是有害的。当然,后面的研究也发现,所谓的X染色体失活也并不完全,确实女性身体里存在一些高于男性的基因表达。但是这些表达的具体影响和相关理论目前尚未完全清楚。
有关X染色体失活理论的发展这里就不展开了,有兴趣的家长可以自行查阅相关资料。总之,女性体内的X染色体一条来自父亲,一条来自母亲,在生长发育过程中,每个细胞中都会随机失活一条X染色体(以及上面的基因)。通常这种失活是随机的,也就是来自父母双方的染色体按50:50(或近似比例)失活。也就是说,对于雷特综合征患者来说,如果是杂合变异,那么大脑里的细胞中应该有一部分是突变的X染色体起作用,另一部分是正常的X染色体起作用,两者比例对半分。
但是,确实有一些女性的X染色体失活是非随机的,甚至是高度非随机(比如,失活比例达到90:10,即来自父亲的X染色体有90%都失活了)。那么,考虑到大部分雷特综合征患者都是来自父亲染色体上的MECP2基因产生了突变[12],来自母亲染色体上的MECP2基因是正常的,如果孩子又恰巧是这种X染色体非随机失活的情况,结果会怎么样呢?显然,在这种情况下正好MECP2基因突变所在的染色体(来自父亲的)大部分都失活了,因而体内就会有更多正常的MECP2基因,最后的表现就会更好一些。当然,反之如果正常MECP2基因所在的染色体大部分都失活了,最后的表现就会更差一些。
这里需要额外说明的是,目前XCI在大部分国内基因检测的时候是不测的。同时,有关体内血液/淋巴液中测的XCI是否能够真实反映脑部细胞,也有待于进一步的研究[6]。
笔者认为,对于家长来说,了解X染色体失活的理论,并不意味着要马上去化验看看自己孩子到底是哪种失活,而更多的还是增强对孩子的信心——基因虽然有突变,但是孩子体内仍然是有正常的基因在工作的,如果通过各种方法刺激这一半正常基因的表达,是能改善孩子症状的。
更多的基因突变类型
前面我们列举了很多基因突变的例子,但是不知道大家是否发现,几乎所有的例子都是发生在外显子(EXON)上的突变。换句话说,我们其实还是只讨论了那2%不到的基因!那发生在那剩余的98%的基因突变,是否就没有问题了呢?
答案是否定的。人们曾经认为那些被裁剪掉的非编码区的基因片段是“垃圾DNA”,认为其不能编码蛋白质,因此是无用的。不过,经过无数科学家们长期的努力,最终人们发现其实DNA中几乎所有的信息都是有用的。虽然它们不能编码蛋白质,但是它们可以“调节”对蛋白质的编码。打个比方,在前面讲到DNA被转录到RNA最终形成mRNA的过程,其所有的操作都不是自然形成的,都需要各种酶来完成。而那些不编码蛋白质的部分,可能会有“吸引”或“排挤”这些酶的功能,从而能加速或减缓基因的表达。
换句话说,即便MECP2基因外显子上没有基因突变,可以正常表达MeCP2蛋白质,但是因为其它部分的基因突变,影响到了基因的转录和/或翻译,从而最终MeCP2蛋白质表达的过多或过少。
我们回到前面Percy的PPT上的例子,3’truncations的意思是在基因的3’端丢失(截断)了一部分基因。虽然这个不影响MeCP2蛋白质相关的碱基,但是这部分的缺失可能会导致,转录过程被抑制,从而还是会缺少MeCP2蛋白质。splice site的意思是在从Pre-mRNA到mRNA过程中,剪切出现了错误(比如少了一个外显子),这样虽然外显子编码没有问题,但是形成的mRNA还是会有问题。某些内含子的基因突变会导致这种情况,让剪切体无法找到正确的位置。
此外,除了MECP2基因外,研究者还发现FOXG1和CDKL5这两个基因的突变也会让患者有雷特综合征相同的症状。不过相对MECP2来说,这两种基因突变更多的是引起非典型雷特综合征。研究分析发现,FOXG1突变更多的引起先天型雷特综合征,而CDKL5突变更多的引起早发癫痫型雷特综合征。不过,和MECP2基因相比,这两个基因的复杂程度要小很多,突变的病例也要少很多。
总之,发生在基因上的任何突变,最终都会对表型上有影响,只是轻重不同而已。当然,总体而言,在非编码区的这些基因突变,其最终的症状还是会相对轻一些,甚至会有很多不显现出来。这一点,也可以从统计中看出来,雷特综合征孩子的基因检测,发生在编码区(也就是这2%区域)的突变远远多于其它区域的。这并非突变在剩余那98%的区域发生的不频繁,而是那些孩子中大部分可能不会有或只有很轻微的雷特症状,从而没有被纳入统计。
到这里,有了这些基础,我们就可以差不多“看懂”孩子的基因检测报告了。
如何阅读基因检测报告
案例解读
前面简要描述基因相关的知识,并大致建立了基因型和表型的关系。那么对我们家长来说,了解这些知识能干什么呢?大部分家长的期望首先就是可以看懂基因检测报告,从而对孩子的病情有一些粗略的了解和对未来的预估。那么,我们先从明白报告中的那些符号开始吧。
首先举一个最常见的例子,这也是包新华(北大医院神经内科)的论文[12]中提到的,在中国雷特综合征患儿中最多的变异:c.502C>T。对于这个变异,通常来说,医院或者检测所给出的基因检测报告是这样的:

这里c.502C>T是什么意思呢?后面的氨基酸变化又是什么意思呢?
先说结论:c.502C>T可以推论出p.Arg168Ter,或者写成p.R168X。换句话说,即便报告中不写p.Arg168Ter,这个信息也可以从c.502C>T推出来。当然,由于这个变异发生在外显子区,通常在讨论归类基因型和表型关系时,多数会用氨基酸来标记。
这里“c.”的意思是突变处于编码区,也就是外显子(用于编码氨基酸)区域,“502”的意思是第502个核苷酸发生突变,“C>T”的意思是原先这里的碱基是C(胞嘧啶),突变称为T(胸腺嘧啶)。
整个编码区有1461个碱基(486个氨基酸加一个终止符 = 487,487×3=1461),其内容如下(来自NCBI,感兴趣的家长可以自己去对比解读一下):

这里一行是100个碱基,从这里可以看到第502个碱基是C(第6行第2个),同时这也是第168个氨基酸对应的第一个碱基,顺序第503、504的两个碱基是G和A,因此原先的排列是CGA,从上面的表格查找可以看到是Arg(简写R)。如果C突变成T,那么排列变成了TGA,从上面表格查找可以看到是Stop(简写X)。因此氨基酸改变可以简写成p.R168X,这里“p.”是蛋白质(protein)的意思,表示这个是用来编码蛋白质的。
再举一个例子:突变c.808delC。这个的意思是编码区第808个碱基C突变缺失了。对应的第808个碱基也是第270个氨基酸的第一个碱基。缺失之后,第270、271个氨基酸原先是CGA(Arg)、AAG(Lys),现在因为C缺失,整体偏移了一个碱基,第270、271个氨基酸变成了GAA(Glu)、AGC(Ser)……一直下去。通常这样的氨基酸变化会记为p.R270fs,意思是原先第270个氨基酸R(Arg)的地方开始发生移位(frame shift)。
按照这种方式,所有的突变都可以类似解读并理解了。有兴趣的家长可以对照孩子的检测报告,计算一下碱基突变对应的氨基酸变化,看看是否和报告中的一致。
对于内含子、5’UTR和3’UTR等区域,因为比较少见,相关的标准记法这里就先略去了。有兴趣的家长可以参考HGVS网页(http://www.hgvs.org/varnomen)。
最后,是里面“纯合/杂合”的意思。“纯合”的意思是一对X染色体上对于的一对等位基因都发生了突变,“杂合”的意思是只有一个染色体上的基因发生了突变。对于MECP2基因变异,几乎都是杂合变异。因此这里大家也可以不用太关心。不过如果是纯合变异,这个案例会非常有学术上的研究价值。
一个奇怪检测报告的解读
上面阐述了如何阅读基因检测报告,那我们再看一个例子,内容是这样的:

说这个例子奇怪,是因为如果根据碱基序列,第799个碱基(第8行倒数第二个)应该是A啊,为什么这里是C呢?
原因很简单,参考碱基不一样,肯定是参考序列不一样造成的。再看参考序列,这里是根据NM_001110792这个序列比对的,而通常是根据NM_004992号进行对比的……那么检测报告错了么?
也没有,只是他们用了不同的参考序列而已。
那么为什么有这么多参考序列呢?这个还是需要回到前面基因表达蛋白质的过程。
在前面基因的表达一节,我们提到了基因中的外显子会经过转录,剪切成为成熟的mRNA,然后在翻译成蛋白质。这里其实还有一些内容没有细说,那就是人类的基因并非每个基因都只有一个剪切方式。实际上,MECP2基因有4个外显子,并不止一种剪切方式。不同的剪切方式会形成蛋白质的不同的同源异构体。具体的,Bird最初发现的MeCP2蛋白质是第2、3、4个外显子的拼接,而这也是NM_004992号对应的异构体(isoform 1)。而NM_001110792对应的异构体是第1、3、4个外显子的拼接(isoform 2),这样其氨基酸序列和编码氨基酸的碱基序列自然不一样了。
不过,这些不同的异构体之间是可以很方便的相互换算的。具体这个例子,我们知道第1个外显子有62个碱基,第2个外显子有26个碱基,那么NM_004992的序列号就是NM_001110792的序列号再往前推36(62-26)号。所以如果按照NM_004992来标记的话,这个突变就是c.763C>T了。对应的氨基酸是CGA突变成TGA,即p.R255X,这是一个常见的突变了。
基因检测报告的用处
如前面所述,通常来说,几乎所有新家长在得知孩子确诊雷特综合征后,都想通过基因突变类型来判断孩子病情的轻重程度和将来的表现。然而,大量的数据分析并没有真正建立起基因型和表型之间的关系。只能说,某几种基因型的突变整体上会“稍好”一些,某几种稍弱一些。但是这都是“整体上”的分析,具体到每一个案例,差别又是很大的。
那么,知道孩子基因突变的类型就一点用没有了么?笔者的想法是,至少有下面两个用处:
1.基因突变点位和表型多少还是有一定关系的。可以通过和其它有同突变点位的孩子交流,找寻共同点,一起研究护理和康复的经验;
2.因为MECP2基因非常复杂,如果将来针对雷特综合征的基因药物出现,其一定是高度个性化的药物。因此,现阶段了解清楚孩子的突变类型,对将来可能出现的药物治疗或者临床实验等,会是一个很好的准备。
目前在国外网上有很多雷特综合征的数据库,比如RettBASE(http://mecp2.chw.edu.au),里面可以查找相关突变点位的所有文献报道,也可以对常见的突变比例进行排序。见下图:
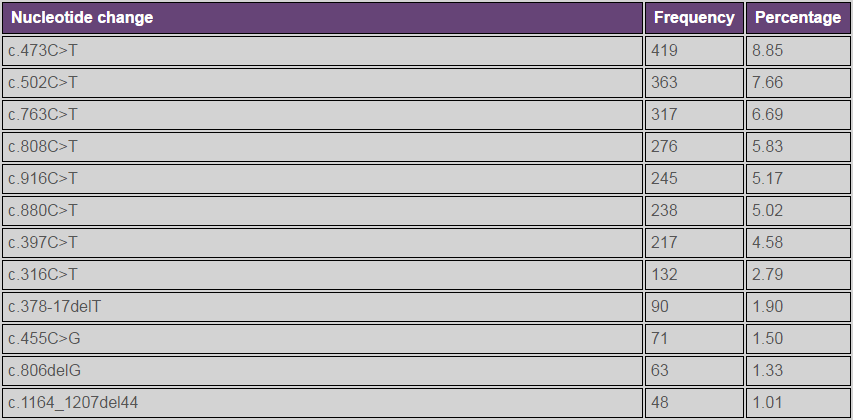
此外,还有一个网上数据库(http://interrett.ichr.uwa.edu.au/output),里面有很多国内外(主要是国外的)研究者和雷特患者家庭多年积攒下来的数据,可以导出基因型和各种症状(例如是否能走路、是否有癫痫、是否有脊柱侧弯等等)关系的数据和图表。里面展现出的相关性会有一定的指导,但是由于雷特综合征患者个体之间差异还是很大,加上东西方生活习惯、饮食习惯和人种基因上还是存在差异,这些数据也只是建议供大家参考。
药物和未来
理论基础
很多雷特家庭,特别是新家长在得知孩子患有这样一种罕见疾病时,都会期待能有灵丹妙药来治好孩子。在网上也不断有各种药物的消息,研究的进展。但是,正如一开头强调的,MECP2是一个非常复杂的基因,雷特综合征是一个非常复杂的疾病,因此对于各种药物和治疗方案,家长们还是应该理性期待。所谓的理性期待,就是大致了解这些药物是怎么一回事,它们是不是真的对自己孩子有效,有多大的效果。
这一章的内容需要有一些基础,建议对雷特综合征的致病原因一章充分理解后再看。
此外,本章只对目前药物研究的原理做一些解释,对于临床相关的知识,请大家仔细看之前安爸的讲座:[安爸和你聊聊研究项目和临床试验,是的,就是讲“药”到底是怎么回事](直播链接:https://m.qlchat.com/topic/details?topicId=220000537001003&isGuide=Y)。
在RSRT(Rett Syndrome Research Trust)的Roadmap to a Cure(https://reverserett.org/cure/)中,用一张图清晰明了的指出了目前关于应对雷特综合征的各种手段:
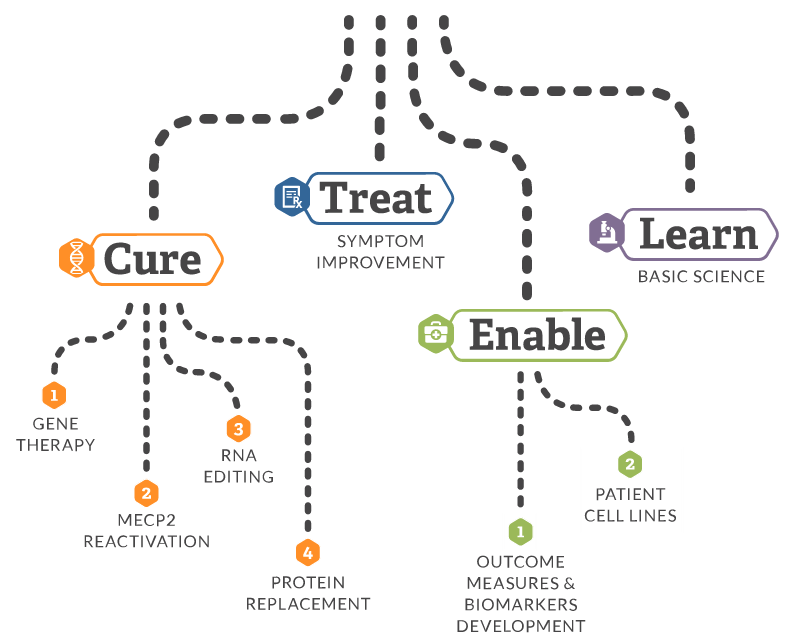
其中Cure(治愈)下面列出了4条路径:
1.基因疗法(Gene Therapy)
2.MECP2再激活(MECP2 Reactivation)
3.RNA编辑(RNA Editing)
4.蛋白质替换(Protein Replacement)
这些目前RSRT推动的几条路径也基本上就是我们能够期待的治愈方式了。
在详细解释这些治疗路径之前,先复习一下相关的理论:
根据前面基因表达、基因突变、以及MECP2相关影响的描述,绝大多数雷特综合征的原因是患者体内一条X染色体上的MECP2基因突变,导致体内正常的MeCP2蛋白质下降到原先的一半左右(根据X染色体失活方式不同而个体有差异),从而影响了其它各种基因的表达。因此,上述的这些不同治疗途径其实就是针对这一整个从基因到蛋白质的链条中的各个环节入手,寻求解决方案:
1.基因疗法 -> 修复突变的MECP2基因,从源头上治愈。
2.MECP2再激活 -> 不修复突变的MECP2基因,而改为激活沉默的那条X染色体上面的MECP2基因,使其得到一定表达来补偿缺少的那部分正常的MECP2基因表达。
3.RNA编辑 -> 从mRNA入手,不去管突变的MECP2基因,而是在基因转录到mRNA过程中通过某种方法得到正确的mRNA,这样就能翻译出正确的MeCP2蛋白质。
4.蛋白质替换 -> 直接提供MeCP2蛋白质到人体内。
从理论上,这些方法都是有可能的。但是从实践上,都存在很多难度。
一个原因是MECP2基因除了复杂外,还有一个特殊的属性。它被称为基因中的金凤花(Goldilocks),其特点是:少了不行,多了也不行(Goldilocks这个名字是来自一个童话故事,大家可以自行搜索)。对于雷特综合征来说,MECP2基因突变导致欠表达,但是还有另外一种罕见病,是突变后X染色体有了2个MECP2基因,称为MECP2重复综合征[13]。这种疾病有重度的自闭症表现,通常以男孩居多。因此,对所有涉及到增加MeCP2蛋白质的治疗方法,必须注意量不能过大,否则可能会引起更严重的后果。而如前面分析,由于X染色体失活和其它基因调节的程度不同,对于每一个雷特综合征患者个例其MECP2基因的问题都不一样,做到适度调节的难度非常大。
另一个原因是人体的血脑屏障(Blood Brain Barrier),这是人体在血浆与脑细胞之间的一种保护机制,可以阻止某些物质(多半是有害的)由血液进入脑组织。血脑屏障随着个体发育的完善而逐步形成,婴儿的血脑屏障功能就远没有成年人强。这也是新生儿会患胆红素脑病(是新生儿黄疸的一种严重并发症)的一个原因:因为新生儿得重症黄疸后,胆红素有可能透过血脑屏障入脑,进而影响中枢神经系统。血脑屏障虽然能帮助大脑抵御外来威胁,但另一方面也阻碍了药物进入大脑。因此在研究针对脑神经药物时,必须要考虑到血脑屏障的影响。
总体来说,方案1(修复突变的MECP2基因)是最理想的,但是其它方案也都有各自的价值,而且各个方案都有科研机构和企业在做这方面的研究。下面会针对这4种途径做进一步的说明。
基因疗法
这种药物可能是所有雷特综合征患者最期望的“指标”的药物。虽然目前为止,这样的药物还没有出现,但是在研究机构和药企中都出现了一些希望。
前面提到的了,2017年作为雷特研究中第4个里程碑,其内容是Bird团队在小鼠身上做的基因修复的实验。虽然这个实验距离真正的应用还比较远,但是基本的技术环节都已经打通了。
让我们来看看这种技术。基因修复的难度有多大?这可跟修房子或者修汽车不一样,也不同于外科手术。大脑中那么多细胞,对每一个细胞中的那么微小的X染色体中的一小片段进行修改,修改还不能损伤别的部位……这肯定不可能通过外科手术的方式来进行。因此,必须要有别的方法。
让我们换一种思路。如果不是单纯考虑“把原来有问题的基因修复”,而是把这个范围扩大一点,考虑“把基因中的部分内容用另外一部分内容替换,最好这种替换能够自发进行,还能从一个细胞传递到下一个细胞”,结果会怎么样呢?
这不就是病毒感染的原理么……
病毒作为一种微生物,出现在地球上的历史比人类长的多。在漫长的进化过程中,病毒形成了非常高效灵活的寄生能力。病毒感染时,就是侵入宿主细胞,把自己的DNA插入/替代宿主的DNA,然后借助宿主的一套能力(前面提到的,DNA -> mRNA -> 蛋白质),复制自己,感染更多细胞等等。
因此,如果找到这样一种病毒,它带有正确的MECP2基因,专门去替代错误的MECP2基因,然后让它去感染雷特综合征患者,那不是就能达到治疗的目的了?
实际上,也确实是按照这个思路做的。目前最常用的是一种腺相关病毒(Adeno-Associated Virus,AAV)AAV9,其特点是穿越血脑屏障到达大脑。Bird团队先通过基因技术制造出来了一批“雷特鼠”,然后以AAV9为载体带上需要替换的DNA,再把这种病毒通过静脉注射到小鼠体内。他们发现,经过这种治疗,小鼠的雷特病征得到了明显的改善。并且,通过对MECP2基因进行裁剪,Bird团队发现如果提供一个“最小”的功能片,就可以有效改善小鼠的雷特病征,从而解决了通常病毒无法携带过长的DNA片段的问题[3]。因此从技术上,这种方式是可行的。
但是,目前的技术到真正能够应用到人体临床上还需要一段时间。首先,小鼠和人类的基因还是有一些区别的;其次,动物实验用的是雄性小鼠,并且小鼠是刻意的敲除了MECP2基因(更具体的说,是去除了EXON2和EXON3的一部分),从而简化了初始情况。对于人类,特别是女孩子来说,如前面所述,大部分是单点的碱基突变,并且存在X染色体失活的影响,因此还需要更多的动物实验(例如在雌性灵长类动物)充分验证后才可能开展人类的临床实验。
目前应用病毒载体修复基因的同类药物已经有一些正在临床。其中比较有名的是AveXis公司用于治疗脊髓性肌萎缩症(Spinal Muscular Atrophy,SMA)I型疾病的AVXS-101[19],根据AveXis公开的资料,药物的效果非常不错,目前正在进行3期临床,有望能快速投入市场。同时,RSRT也在和AveXis合作准备应用相同的技术开发治疗雷特综合征的药物。
MECP2再激活
根据RSRT的信息,目前这种方案主要是在一些研究机构做理论研究。各个机构在用不同的方式和不同的实验对象在探索让沉默的X染色体上的部分基因得到表达的方法。不过迄今尚未有能用于临床的技术出现。
RNA编辑
根据RSRT的信息,目前这种方案也主要是在研究机构进行。RNA编辑的基本思想是从剪切体入手,“嫁接”正确的Pre-mRNA部分,替换掉MECP2基因突变部分转录的Pre-mRNA,如下图[14]:
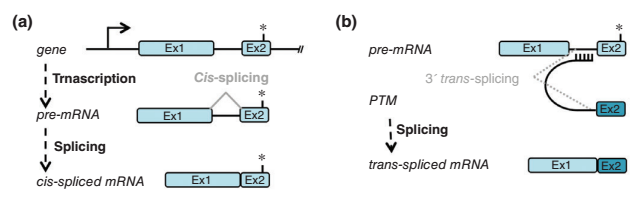
这里(a)是正常的基因表达途径,基因有两个外显子(这里用Ex1,Ex2表示),其中Ex2存在缺陷。这样最后得到的mRNA就会带有这个缺陷。(b)是RNA编辑的思路,在细胞内提供含正确Ex2信息的PTM(pre-mRNA trans-splicing molecule)来替代掉缺陷的Ex2,从而在剪切完成后得到的是正确的mRNA,进而能表达出正常的蛋白质。
是目前看来,这也是一种非常有前途的治疗基因缺陷疾病的途径。
蛋白质替换
这条途径也是很自然的一种思路:既然雷特综合征患者缺少这个MeCP2蛋白质,那我们人工补上不就可以了吗?然而“补”并不简单,必须要克服“如何补”(突破血脑屏障)和“补多少”(控制剂量,避免出现MECP2重复综合征的症状)。相比起其它几种途径,从蛋白质入手也有自己的优势。因为尽管过多的MeCP2会导致MECP2重复综合征,但是对于雷特综合征这样已经长期缺乏MeCP2的个体,是否初期用药时“矫枉过正”一下反而会取得更好的疗效呢?其它方案从原理上只能达到正常MeCP2的目标,只有蛋白质这条方案可以实现短期过量补充。因此从这个角度上,蛋白质这条途径有它自己的优势。
在这条道路上,突破血脑屏障是一个重要的技术。目前有一家叫ArmaGen的公司已完成两种药物AGT-181和AGT-182,分别针对黏多糖病I型和II型,现在都处于临床试验阶段。同时,ArmaGen也得到RSRT的资助,在研发针对雷特综合征以及其它一些神经系统疾病的药物。
当前已有/正在做临床的药物
前面描述的Cure(治愈)路径下的药物都还停留在实验室阶段,尚未有任何一种针对雷特综合征的治愈药物进入临床。但是,对家长而言,并非只有能治愈的药物才有价值。在Treat(治疗)下面,注重改善症状的药物其实对现阶段的孩子也是有益的。
这类药物的思路基本上都是从症状入手,因为已知有很多疾病的部分症状是和雷特综合征类似的,那么很自然的就会想到:对应这些疾病症状的药品是否也能用于雷特综合征呢?当然由于病因的不同,在其它疾病上比较有效的药物可能不一定对雷特综合征有效,因此有关这类药物其实一直在进行临床研究,在医院诊断时医生也只能是给出一些建议,对于具体每一个雷特患者个体只能是在保证安全性的前提下进行一些尝试而已。
需要说明的是,这些目标是改善症状的药物目前都多多少少进行了临床研究。而临床试验的结果其实都比较一般,绝大多数药物虽然能在某几个指标上验证有效,但都没有能证明在雷特综合征严重度指标上的改善。这其实并不意外,因为本身雷特综合征就是非常复杂的疾病,患者个体差异也很大,如果不从基因角度入手的话,很难取得一致的结果。因此,所谓“某某药临床试验失败”,并不意味着这款药就一无是处。如果调整一下临床试验方案,对入排标准起点终点等进行细化,或许这款药能够应对一部分雷特综合征患者的症状,改善她们的生存状况。
下面列出一些有关这类改善症状的药物,在论文上和https://clinicaltrials.gov/上都可以找到记录。这些药物的相关研究也一直在进行,有兴趣的家长可以关注一下。
NNZ-2566
相信很多家长都听过NNZ-2566(现在叫Trofinetide)的名字。这个药来自Neuren这家公司,应该说是目前临床进展最快的用于治疗雷特综合征的药物了。这个药的思路是,Neuren发现雷特综合征患者有脑损伤的表现,那么不管这个脑损伤是MECP2基因突变导致的还是别的什么导致的,只需要“修复”脑损伤,患者的病情就能有好转。
基于这个思路,Neuren研究了IGF-1生长因子(IGF-1可以有效促进大脑发育),并进行最终研发出了Trofinetide这个药。有关Trofinetide的原理,有兴趣的家长可以阅读Neuren网站上的介绍[15]。简单来说,就是化学合成提供了一种有部分IGF-1生长因子的药物,其相比生物IGF-1更加易于保存且可以通过口服吸收,在实践中更加方便。根据Neuren提供的资料,动物实验表明Trofinetide可以帮助大脑中神经元的树突发育,从而提升大脑的功能。
所以,NNZ-2566并非专门针对雷特综合征研发的药物,还可以适用于诸如X脆性染色体综合征等基因突变导致的遗传病,以及包括阿尔兹海默症(老年痴呆)、帕金森综合征等在内的脑损伤疾病。
根据最新公开的信息,Trofinetide在治疗雷特综合征的临床上已经完成了2期试验。后面将会进行3期临床试验。但是从原理上,这个药并不能完全“治好”雷特综合征,只是可能相比起其它药物会更加能改善患者的症状。
左旋肉碱
早在2000年左右的时候,就有应用左旋肉碱(L-carnitine)来改善雷特综合征的临床研究[16]。国内北大一院包新华教授也曾经进行过用左旋肉碱的研究[17]。但是总体来说,左旋肉碱对症状的改善一说缺乏统计学意义,因此后来并未进行大规模的研究。当然,也有一种理论说,左旋肉碱对于生酮作用是有帮助的,而酮环境对于癫痫控制是有利的(有一些幼儿顽固性癫痫可以通过生酮饮食来改善)。也有说一些抗癫痫的药物可能会有一些副作用,但是补充左旋肉碱可以在代谢途径上给予辅助,降低副作用[18]。不过总体来说,左旋肉碱只能说对部分雷特综合征患者会有用。
笔者认为,左旋肉碱本身作为一种营养品,吃了也不会有什么坏处。但是也不会有什么本质上的改善。吃左旋肉碱的考虑,可能就跟吃DHA、钙镁液等差不多,带着一种吃营养品的心态吃就好了。
右美沙芬
右美沙芬是一种常用的止咳药,在感冒咳嗽时去医院医生很多时候也会开这种药来缓解咳嗽症状。看上去这个药和雷特综合征似乎没有什么关系,但是研究者发现,右美沙芬是N-甲基-D-天门冬氨酸受体(NDMA受体)拮抗剂,而NDMA受体有自己奇特的药理性,可以调节神经元的存活,调节神经元的树突、轴突结构发育及参与突触可塑性的形成,甚至神经元回路的形成。研究者进行了用右美沙芬改善雷特综合征患者的临床试验,结果发现不错:首先右美沙芬对于雷特患者是安全的,其次虽然整体雷特症状严重度指标上没有看到明显改善,但是在具体一些指标,包括癫痫、语言理解、行为活跃度上是有改善的[20]。
不过右美沙芬目前只是完成了第一期的临床,样本量也有限。还需要进一步的临床研究。
EPI-743
EPI-743的通用名称是Vatiquinone,是Edison Pharmaceuticals研发的治疗能量代谢疾病的药物。这款药物取得了FDA授予的治疗Leigh氏综合征的孤儿药地位。Leigh氏综合征是一种亚急性的疾病,患者常常发病后经数周或数月后死亡。临床症状包括肌张力障碍,痉挛,癫痫等,部分也有智力障碍。
EPI-743是从代谢入手,其药理上可以提高谷胱甘肽的合成并优化代谢控制,从而改善症状。应用在Leigh氏综合征的临床上表现不错。Edison也进行了在包括雷特综合征在内的多种神经系统疾病上的临床试验。特别的,应用在雷特综合征上已完成了II期临床试验,不过临床未能达到目标。后面是否会调整临床试验方案继续试验也还没有决定。
UX007
UX007的正式名称叫Triheptanoin,是由Ultragenyx公司研发的一种合成甘油三酯化合物。UX007是为1型葡萄糖转运蛋白缺乏综合征(Glut1 DS)研发的,这种疾病的发病症状包括癫痫、发育迟缓和运动障碍等,目前也没有FDA批准可用的药物。
UX007是从代谢途径入手,当人体吸收这种药物后,可以通过代谢给患者提供硬脂酸瘐酸酯,而这种物质可以通过血脑屏障,最终转化为葡萄糖给大脑提供能量[21]。在临床研究中,UX007已经完成了在Glut1 DS上的二期临床,还在继续研究。
由于雷特综合征和Glut1 DS的部分症状类似,因此也有将UX007用于雷特综合征的想法。不过由于各种因素,目前I期临床研究还尚未开始。
各种抗癫痫药物
由于目前还没有完全清楚部分雷特综合征患者出现癫痫症状的原因,因此对于癫痫来说只能采取“治标”的方法来处理。研究表明,常用的抗癫痫药物对于出现癫痫的雷特综合征患者也是有效的,但是每一个个例都不一样,可能也需要尝试才能够找到合适的药物。具体药物的选择都是根据临床表现、病史和其它生化指标定的,还是需要咨询经验丰富的医生。同时也需要定期随访跟踪,才能控制好。
其它针对神经系统疾病的药物
由于雷特综合征患者的一些表现和精神类疾病类似,因此研究者也进行了相关药物应用在雷特综合征上的临床研究。这些药物包括针对精神分裂症的沙立佐坦(Sarizotan)、利培酮(Risperidone)等,以及针对多发性硬化症(MS)的芬戈莫德(Fingolimod)、醋酸格拉替雷(Copaxone)及其仿制药Glatopa等。这些药物都完成了一些临床试验,但是目前尚无进一步的消息。
如何期待
对于治疗雷特综合征的药物,目前看确实是有希望的,只是具体要多久才能临床应用尚不清楚。同时,如前面所述,MECP2基因非常的复杂且挑剔,未来的药物治疗很有可能会是个性化的药物。对治愈(Cure)类药物和治疗(Treat)类药物都是如此。因此,各位家长熟悉自己孩子的病情,掌握孩子生长发育的数据,对于将来可能出现的临床试验也好,实际临床应用也好,都会有很大帮助。
此外,对于针灸、中药等治疗方式,目前从原理上无法证实其对MECP2基因表达或者其上下游基因表达有效,同时也还没有证据表明这些治疗方式对雷特综合征有什么直接效果。
笔者个人认为,从科学性和安全性上说,与其寄希望于针灸和中药,还不如把钱和时间放到康复训练(即前面提到的环境影响),因为这些是经过一些实践有可能提高某些基因的表达,进而改善或者缓解症状的。
总结
让我们回到最开头。我们回顾了雷特综合征的研究历史,并结合现代基因理论对雷特综合征的致病原因做了解释。最后按照RSRT上给出的治愈/治疗路径和https://clinicaltrials.gov/上面的数据列出了当前对药物的研究状况。总体来说,雷特综合征及其相关的研究一直在不断进行,不管研究机构还是企业都有很多人在为此努力。
笔者个人认为,雷特综合征的历史上每约17年会有1个里程碑,今年(2017年)是第4个里程碑,那么第5个里程碑或许就出现在17年之后(2034年),而其内容将是雷特综合征这一罕见病得到完全的解决。
参考文献
1.Amir R E, Veyver I B V D, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encodingmethyl-CpG-binding protein 2[J]. Nature Genetics, 1999, 23(2):185.
2.Chen Y, Yu J, Niu Y, et al. Modeling Rett Syndrome Using TALEN-Edited MECP2 Mutant Cynomolgus Monkeys[J]. Cell, 2017, 169(5):945.
3.Tillotson R, Selfridge J, Koerner M V, et al. Radically truncated MeCP2 rescues Rett syndrome-like neurological defects.[J]. Nature, 2017, 550(7676):398.
4.Nan X, Campoy F J, Bird A. MeCP2 Is a Transcriptional Repressor with Abundant Binding Sites in Genomic Chromatin[J]. Cell, 1997, 88(4):471.
5.Suter B, Treadwell-Deering D, Zoghbi H Y, et al. MECP2 Mutations in People without Rett Syndrome[J]. Journal of Autism & Developmental Disorders, 2014, 44(3):703.
6.Amir R E, Van D V I B, Schultz R, et al. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes[J]. Annals of Neurology, 2000, 47(5):670-9.
7.Ehrhart F, Coort S L M, Cirillo E, et al. Rett syndrome – biological pathways leading from MECP2 to disorder phenotypes[J]. Orphanet Journal of Rare Diseases, 2016, 11(1):158.
8.Kwan H C, Li H H, Charandle J, et al. Cerebellar gene expression profiles of mouse models for Rett syndrome reveal novel MeCP2 targets[J]. BMC Medical Genetics,8,1(2007-06-20), 2007, 8(1):1-16.
9.Nan X, Meehan R R, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2.[J]. Nucleic Acids Research, 1993, 21(21):4886-92.
10.Cuddapah V A, Pillai R B, Shekar K V, et al. Methyl-CpG-binding protein 2 (MEPC2) mutation type is associated with disease severity in Rett Syndrome[J]. Journal of Medical Genetics, 2014, 51(3):152-8.
11.De B C, Zappella M G, Meloni I, et al. Preserved speech variant is allelic of classic Rett syndrome.[J]. European Journal of Human Genetics, 2000, 8(5):325-330.
12.张晓英, 赵滢, 包新华,等. 中国人群Rett综合征的遗传特点与机制研究[J]. 中华医学遗传学杂志, 2014, 31(1):1-5.
13.Esch H V. MECP2 Duplication Syndrome - GeneReviews® - NCBI Bookshelf[J]. Gene, 2010.
14.Berger A, Maire S, Gaillard M C, et al. mRNAtrans‐splicing in gene therapy for genetic diseases:[J]. Wiley Interdisciplinary Reviews Rna, 2016, 7(4):487.
15.http://www.neurenpharma.com/irm/content/the-science-behind-neuren-s-products1.aspx?RID=304
16.Ellaway C J, Peat J, Williams K, et al. Medium-term open label trial of L-carnitine in Rett syndrome.[J]. Brain & Development, 2001, 23(1):S85-S89.
17.包新华, 潘虹, 宋福英,等. Rett综合征的临床特征及MeCP2的基因型与表型的关系研究[J]. 中华儿科杂志, 2004, 42(4):252-255.
18.陈蕊, 张明. 丙戊酸相关肝毒性的治疗进展[C]// 华东地区脑电图与临床神经电生理学术会议——暨江西省抗癫痫协会年会. 2014.
20.Smithhicks C L, Gupta S, Ewen J B, et al. Randomized open-label trial of dextromethorphan in Rett syndrome.[J]. Neurology, 2017.
22.Beall C M, Ellison P T. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders.[J]. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(25):11459.
原文作者:上海丫丫爸
备注:安爸

